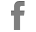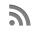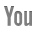Gardner’s syndrome is an autosomal dominant disease characterized by the presence of colonic polyposis, osteomas and a multitude of soft tissue tumors. The syndrome may present at any age from 2 mo to 70 years with a variety of symptoms, either colonic or extracolonic. We present a case of a 11-year-old female patient with Gardner’s syndrome who presented with a lumbar area desmoid tumor and treated with resection of the desmoid, restorative proctocolectomy and ileal pouch anal anastomosis, A review of the current literature has been performed.
INTRODUCTION
Gardner’s syndrome is a rare (1:1 000 000 population in USA) autosomal dominant inherited disorder with a high degree of penetrance characterized by the triad of colonic polyposis, multiple osteomas and mesenchymal tumors of the skin and soft tissues[1-3].
In 1951, Gardner reported the association between surface tumors and colonic polyps that are prone to malignant degeneration[4]. In 1952, Gardner and Plenk described the dominant hereditary pattern of multiple osteomas associated with colonic polyposis[5]. Later that year, Gardner and Richards reported the association of hereditary colonic polyposis and osteomatosis with multiple cutaneous and subcutaneous tumors describing the so-called Gardner’s syndrome[6].
It is believed that Gardner’s syndrome and familial adenomatous polyposis (FAP) are variants of the same disorder, since they share the same genetic alterations. The fact that Gardner’s syndrome is associated with extracolonic manifestations may be explained by a variable penetrance of a common mutation. The disorder is linked to band 5q21-q22, the adenomatous polyposis coli locus (APC gene). More than 1 400 different mutations of this gene have been reported. The specific area of the APC gene that is mutated determines the extracolonic manifestations as well as the number, time frame and malignant potential of adenomatous polyps. Although most cases show familial clustering, one-third of cases occur due to spontaneous mutations. MYH (1p34.3-p32.1) is another gene associated with FAP. In general, the clinical picture of FAP patients with mutations in the MYH gene is milder than those with mutations in the APC gene. The presence of extracolonic manifestations is linked to the vast majority of cases, if not exclusively, to mutations of the APC gene. It is believed that in the pathogenesis of FAP and possibly Gardner’s syndrome, environmental factors such as diet, exercise and smoking also play an important role[1,7-11].
The clinical spectrum of disease presentation is variable and often diagnosis is delayed, despite the presence of clues for a significant amount of time. We present the rare case of a 11-year-old girl with Gardner’s syndrome that was diagnosed due to the finding of a desmoid tumor on the lumbar area.
CASE REPORT
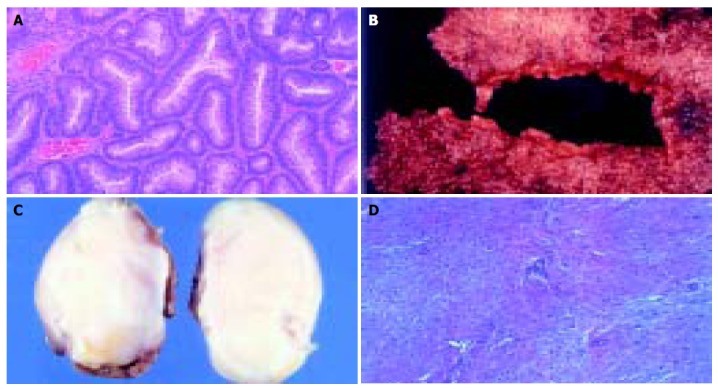
A 11-year-old female patient presented with a small tumor in the right lumbar area. The patient was afebrile with normal vital signs. Giordano’s sign was absent and she had a normal urinalysis, CBC and blood biochemistry. After careful clinical examination, the only abnormal finding was a 4-cm mass on the right lumbar area. The mass was fixed, non-tender and had a bony firm consistency. IVP was scheduled with insignificant findings. Follow up of the tumor growth was advocated for a year before further investigations should take place, but the rapid increase in tumor size lead to a CT scan evaluation of the abdomen and pelvis 3 mo later. A 6-cm tumor of indeterminate quality was revealed originating or adhering to the left latissimus dorsi muscle. After careful medical history was taken again, the patient recalled an episode of hematochezia that had been attributed to hemorrhoids. On colonoscopy, she was found to have numerous polyps carpeting the entire colon and rectum, which was consistent with FAP. The clinical suspicion of Gardner’s syndrome was raised. No family history of FAP was evident. A fundoscopic examination was performed showing the typical hypertrophy of the pigmented layer of the retina. An upper endoscopy was performed revealing normal findings. Subsequently resection of the lumbar tumor was performed via a posterior approach. The pathologist reported a firm tumor with a gritty sensation during transection. On cross section, a glistening white coarsely trabeculated surface was revealed resembling scar tissue (Figure 1A). On microscopic examination, the tumor consisted of eosinophilic or amphophilic spindle-shaped cells embedded within moderate amount of collagen. These cells did not show atypical or hyperchromatic nuclei but some pleomorphism was present (Figure 1B). The final report was that of a desmoid tumor. The next step was a restorative proctocolectomy with ileal pouch anal anastomosis (RPC/IPAA) and mucosectomy. Pathology of the specimen confirmed the diagnosis of FAP (Figure 1C). Colonic polyps proved to be tubular adenomas. The epithelial cells showed moderate differentiation into normal cells although scattered goblet cells could be found. Cellular atypia were manifested by tall cells with pseudostratified nuclei (Figure 1D). No malignant transformation was present.
On discharge, it was recommended that all family members must be evaluated for FAP. Still, the patient was lost to follow-up 2 mo later because of address change.
DISCUSSION
Gardner’s syndrome represents a multisystemic disease. Symptoms are usually evident by the 20th year of age, but they may present anytime between 2 mo and 70 years. In general the cutaneous and bone abnormalities develop approximately 10 years prior to polyposis[1,3,12]. However, in our case the polyps were already symptomatic at the time of diagnosis, before the extracolonic manifestation of the disease became evident.
The gastrointestinal manifestations of Gardner’s syndrome include colonic adenomatous polyps (tubular, villous, tubulovillous), gastric and small intestinal adenomatous polyps (12% of patients) and peri-ampullary carcinomas (2% of patients). Polyp formation starts at puberty but diagnosis is usually in the third decade, while malignant transformation approaches 100% by the fourth decade of life. The presenting symptoms are anemia, lower gastrointestinal bleeding, cramping abdominal pain, diarrhea, bowel obstruction and mucous discharge. Most of the polyps are small (<5 mm) and therefore difficult to demonstrate on imaging studies. The larger sized polypi may prolapse through the anus or may lead to intussu-sception or ileus. On colonoscopy, the rectum and whole large intestine appear carpeted by the polyps[1,3,12-14].
Osteomas appear in about half of Gardner’s syndrome patients and manifest earlier than polyposis. They may be endosteal or exosteal and demonstrate sclerosis and deformity. The vast majority of osteomas are located on the skull. Since the most commonly affected bones are the mandible and maxilla, many patients present initially to dental surgeons. However, long bones and even phalanges may be affected. A finding of three or more osteomas has been proposed as a screening method for Gardner’s syndrome[1,12,15].
The commonest skin manifestations of Gardner’s syndrome are epidermoid or sebaceous cysts (66%) and are found on the face, scalp and extremities. They are usually asymptomatic although mild pruritus or signs of inflammation may be evident. Other skin manifestations are fibromas, neurofibromas, lipomas, leiomyomas, and pigmented skin lesions[1,16].
Desmoid tumors appear in 3.5-5.7% of patients and usually appear within 3 years following surgery although they can appear at anytime. Common locations are the incision sites, the abdominal cavity (especially the mesentery) or the retroperitoneum. They are considered as one of the most troublesome manifestations of Gardner’s syndrome since they may cause life threatening complications and are usually resistant to treatment[1,17-22]. In our case, the desmoid tumor was the sole manifestation of the disease and with a misleading medical history, diseases of the left kidney and ureter were first considered.
In about 90% of population, hypertrophy of the retinal pigmented layer occurs. Other manifestations of Gardner’s syndrome are papillary thyroid cancer (often multicentric), benign intracranial neoplasms such as meningiomas and epidermoid cysts, hepatoma, hepatoblastoma, fibromas, leiomyomas, lipomas, biliary and adrenal neoplasms, osteosarcoma, chondrosarcoma, and in 70% of patients dental disorders (congenitally missing teeth, hypercementosis, odontomas, dentigerous cysts, impacted teeth, supern-umerary teeth, fused or unusually long roots and multiple caries)[1-9,12-16].
The diagnosis of FAP and Gardner’s syndrome can be made by genetic testing for gene mutations or by the demonstration of multiple colonic polyps during colonoscopy. Genetic testing is the most effective method for demonstrating mutated APC gene in relatives of patients with FAP syn-drome[1,23].
Owing to the increased risk of cancer in various organs, patients with FAP/Gardner’s syndrome or their relatives should undergo surveillance programs. These include both large bowel and upper GIT surveillance, as well as thyroid and possibly hepatic surveillance too.
Large bowel surveillance is indicated in the following cases:
I Family history of FAP, but no demonstrable mutation
Annual flexible sigmoidoscopy starting from the age of 13-15 till the age of 30. Sigmoidoscopy every 3-5 years from 30 to 60 years.
II Known APC mutation, but patient refuse surgical treatment.
Flexible sigmoidoscopy every 6 mo and annual colonoscopy starting from the age of 10-12. Advise surgery at an age, earlier than 25 years.
III Known FAP following surgery Annual rectoscopy following ileo-rectal anastomosis (IRA).
Annual proctoscopy following IPAA.
Today no widely accepted upper gastrointestinal tract surveillance program exists. However, all patients with known FAP should undergo an upper GIT surveillance program, due to higher risk of malignancy development in this area. A triennial upper gastrointestinal endoscopy utilizing both direct and side view endoscope has to be performed. In the case of multiple polyps, annual endoscopy is advised.
Annual clinical and ultrasonographic examination of the thyroid gland is mandatory. Hepatic surveillance by ultrasound and AFP level measurement is under investigation[1,23].
Treatment is advised at diagnosis considering the malignant potential of the polyps and due to the fact that 25% of cases presenting with symptoms have cancer. Treatment options include total proctocolectomy and permanent terminal ileostomy, RPC and construction of an IPAA, subtotal colectomy and IRA. Prophylactic resection should be performed before the 25th year of life, ideally between the age of 16 and 20 years. However, patients with a large number of polyps early in life may benefit from an operation at an earlier age, considering the relationship between polyp number and cancer risk. None of these options is ideal and the choice of operation is always a combination of the patient’s preference and the surgeon’s experience. The first operative option seems to be the safest, since no large intestinal epithelium remains, but the patient must learn to live with an ileostomy. The second option avoids an ileostomy, but is more technically demanding, is associated with greater morbidity and the need for lifetime follow-up, due to the possibility of residual mucosa. The third option should be reserved for those patients whose rectum is relatively disease free (less than 20 polyps). Once thought to result in spontaneous regression of the remaining rectal polyps, this procedure appears to be of questionable efficacy. In our case, we performed the more technically demanding RPC/IPAA after explaining to the patient (and her relatives) the possible choices[1,3,11,24,25].
Owing to the morbidity and mortality associated with the surgical treatment options, there is intensive investigation for drugs that could decrease the malignant potential of adenomatous polyps. In 1983, Waddell and Loughry reported that the rectal polyps of patients with FAP almost disappeared following sulindac administration. Sulindac and other NSAID’s have been tested in subsequent clinical studies demonstrating a reduction in the number and size of polyps during the period of their administration. However, the appearance of new polyps as well as the malignant transformation of polyps has been demonstrated in patients on sulindac therapy. In order to avoid the side effects of long-term NSAID administration while maintaining efficacy, researchers have focused on the newer COX-2 inhibitors (celecoxib, rofecoxib) following the demonstration of COX-2 expression in colonic carcinomatous mucosa but not in normal GIT mucosa. A decrease in the polyp size was noted but at higher than usual doses. Currently celecoxib 400 mg/d has granted FDA approval for FAP patients by decreasing the polyps’ size, but whether it reduces the malignant potential of them remains to be investigated[11,26].
Non-NSAID substances that have been tested for the prevention of malignant transformation of polyps in FAP patients seem to be of inferior efficacy. These include 5-FU, Vit C, Vit E, calcium, DHA and green tea extract[11].
Treatment options for the cutaneous cysts of Gardner’s syndrome are the same as for ordinary cysts. Treatment is indicated for symptomatic cases or for cosmetic reasons. In the same manner, osteomas may require resection if they interfere with function or for cosmetic reasons[1,16].
Treatment of desmoids is generally considered to be a challenge for both the doctor and the patient. Operation is recommended only for symptomatic patients due to common recurrence (up to 80%). Medical treatment consists of sulindac, tamoxifen or toremifene, raloxifene alone or in combination. Doxorubicin, dacarbazine as well as carboplatin, vinblastine and methotrexate have been used in case of aggressive unresectable desmoids. Radiotherapy can also be tried. The treatment plan greatly depends on the location of the desmoid tumor. Mesenteric desmoids tend to involve the mesentery diffusely, altering gross anatomy and can invade vessels or ureters. Often complex operative procedures have to be employed and recurrence is typical. So a less aggressive medical approach is preferred initially and surgery should be performed in case of failure. Abdominal wall desmoids should be treated surgically since they are easier to be excised and recurrence rates are lower, compared to their mesenteric counterparts[1,17-22]. In our case, we had an unusual occurrence of a desmoid tumor at the muscles of the back. The excisional biopsy raised the clinical suspicion and led to the final diagnosis and treatment. Still, there are no follow-up data to evaluate recurrence.
In conclusion, we presented a case of Gardner’s syndrome with an unusual clinical presentation of a desmoid tumor in the left lumbar area that led to the final diagnosis after taking careful medical history. RPC/IPAA was the surgical modality of choice with excellent immediate post-operative results. Even though in our case the patient was lost to follow-up it cannot be over expressed that after the diagnosis of Gardner’s syndrome, the patients must be aggressively followed-up, since there is a constant threat to their lives at any age.
References